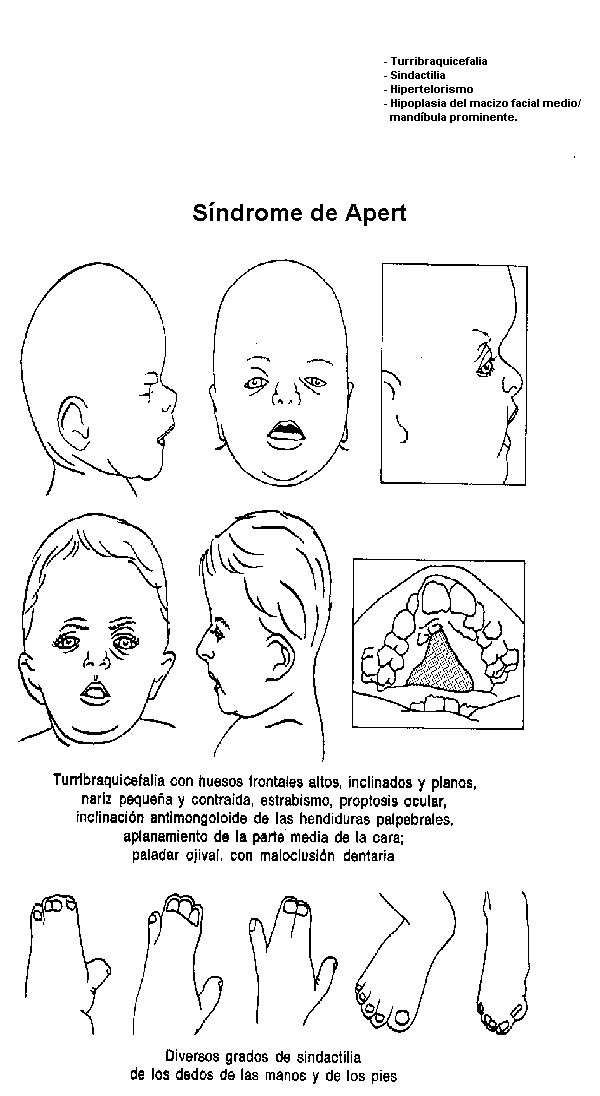
Características clínicas. El
síndrome de Apert se caracteriza por craneosinostosis congénita
que
conduce a una turribraquicefalia, sindactilia de las manos y de los
pies, anquilosis diversas y sinostosis progresiva de las manos, de los
pies y de la columna vertebral. La mayoría de los pacientes que
presentan el síndrome son retrasados mentales. Se observa casi
siempre la existencia de acné vulgar, la cual suele propagarse
a los antebrazos. Es muy notable la variabilidad de las facciones. Las
órbitas son muy hipertelóricas. El macizo. facial medio
suele estar hipodesarrollado, lo cual presta prominencia a la mandíbula.
El cráneo está malformado, con aplanamiento de los huesos
frontal y occipital, y el vértice del cráneo localizado
en las proximidades del sincipucio, o en su parte anterior. Es frecuente
el cierre precoz e irregular de las suturas craneales, especialmente
la coronal. Las impresiones digitales están a menudo aumentadas.
El paladar es ojival y puede tener un surco medio. En una tercera parte
de los casos se ha observado una fisura palatina en el paladar blando.
Es frecuente la maloclusión. debido ala hipoplasia de la parte
media de la cara.
Diagnóstico específico. Las
manos y los pies están deformados de una forma simétrica.
Se halla una
masa a nivel medio de la mano, con sindactilia ósea y de los
tejidos blandos de los dedos II, III y IV. A menudo, los dedos I y V
se hallan unidos de una forma incompleta a la masa de la parte media
de la presentan una completa sindactilia de los tejidos blandos, y que
generalmente tienen una uña común. En varios casos se
ha observado la presencia de 6 metatarsianos. Con la edad, las articulaciones
interfalángicas de los dedos de las manos se hacen rígidas.
Existe un acortamiento de las extremidades superiores, y puede haber
anquilosis de las articulaciones, especialmente del codo, del hombro
y de las caderas. Los huesos de las manos, de los pies y de la columna
vertebral, van
formando sinostosis progresivamente.
Diagnóstico diferencial. El trastorno
se debe diferenciar del síndrome de Pfeiffer, del síndrome
de
Crouzon y del síndrome de Carpenter. Es probable que el síndrome
de Summitt sea idéntico al
síndrome de Carpenter. Una sindactilia acentuada, como la que
se presenta en el síndrome de Apert, puede observarse en el síndrome
de criptoftalmos.
Diagnóstico prenatal. Se ha efectuado
el diagnóstico prenatal, al observar por fetoscopia las
alteraciones de las manos y de los pies.
Defecto básico, genética y
otras consideraciones. No se conoce el defecto básico. Existe
una
herencia autosómica dominante. Se ha observado una edad paterna
avanzada. Aunque la mayoría de los casos son esporádicos,
existen varios ejemplos en los cuales una mujer afecta ha dado a luz
a
un niño en el que aparecía el síndrome. Se han
publicado más de 250 casos. Se calcula que se
presenta aproximadamente en 1 por cada 160.000 nacimientos. Sin embargo,
debido al elevado
porcentaje de mortalidad que ocurre en el período neonatal, existe
aproximadamente 1 caso por cada 2.000.000 en la población general.
No parece existir heterogenicidad genética.
Pronóstico y tratamiento. El pronóstico
depende en gran parte del grado de retraso mental existente. Aunque
habitualmente no se efectúa tratamiento alguno para la corrección
de la sindactilia de los dedos de los pies, generalmente se llevan a
cabo considerable número de intervenciones sobre la sindactilia
de las manos, con el fin de conseguir la superación del pulgar
y del meñique del resto de los dedos, y mejorar así la
aprehensión. Se pueden efectuar importantes intervenciones de
cirugía maxilofacial para la corrección de la deformidad
del macizo facial medio, con el propósito de llevar el maxilar
superior hacia delante, así como corregir el exoftalmos.
Autor: Goodman, R
Titol: Malformaciones en el lactante i en el niño.
Editorial Salvat.