|
|
LA
TALASSÈMIA
Què és la talassèmia?
Són un grup heterogeni de trastorns
clínics que tenen en comú un defecte constitucional
d’hemoglobinització dels eritròcits,
i inclou des de casos realment greus fins a casos en què els
individus tan sols són portadors asimptomàtics.
Són malalties hereditàries
de la sang que inclouen defectes en la hemoglobina, component
dels glòbuls vermells encarregat de transportar
l’oxigen. En cada molècula d’hemoglobina
hi ha dos tipus principals de proteïnes anomenades
globines, que segons l’etapa de la vida en que es
troba una persona seran d’un tipus o bé d’un
altre, essent les més importants la globina alfa
i la globina beta. Els individus que pateixen talassèmia,
no produeixen una d’aquestes proteïnes (i en
ocasions cap de les dues) o bé no les sintetitzen
amb una qualitat suficient per a ser prou eficients per
a que l’organisme les faci anar. Com a resultat,
els seus glòbuls vermells solen ser anormals i no
estan en condicions de dur a terme les seves funcions.
Origen de la malaltia...
La talassèmia va ser descrita per
primera vegada per Cooley i Lee el 1925 en pacients nens
d’origen italià amb anèmies severes
i engrossiment de fetge i melsa, pal·lidesa i canvis
en els ossos.
Per altra banda, el terme “thalassèmia” no
va ser usat fins a set anys desprès de la seva primera
descripció, quan Whipple i Bradford van publicar
un document descrivint la patologia de la malaltia.
Etimològicament, el terme “thalassèmia”prové de
la unió de dues paraules gregues; mar (тŋαλασσα thalassa)
i sang (αıμα - aima); que
en conjunt ve a significar “mar a la sang” o “anèmia
del mar”. Amb aquesta denominació, George
Whipple va voler donar a entendre que aquesta malaltia
mostrava especial predilecció per les poblacions
que habitaven vora el mar, en el nostre cas, el mar Mediterrani.
Distribució geogràfica
de la talassèmia |
Una ullada a la distribució global de les talassèmies
ens demostra que es produeixen en major quantitat a la
regió mediterrània, on es van detectar
primer. No obstant, el temps ha demostrat que aquesta
malaltia no és exclusiva de l’àrea
mediterrània, sinó que afecta també a
individus de raça àrab, africans, indis,
del sud-estasiàtic i de l’Orient Llunyà.
En aquesta regió, són una causa significativa
de mortalitat, però són freqüents
perquè les persones heterozigòtiques estan
protegits de la infecció severa del paludisme.
La malària és provocada pel paràsit Plasmodium
falciporum a través de la picada del mosquit Anopheles. Aquest
paràsit, roman part del seu cicle vital en els
eritròcits i s’alimenta de l’hemoglobina,
però els malalts heterozigotics amb talassèmia,
especialment la beta, són en certa manera immunes
a aquesta malaltia. Aquesta és la raó per
la qual en els països on encara hi ha brots de malària,
gran quantitat dels seus habitants són beta-talassèmics.

Imatge
16: Mosquit
transmissor de la malària.
www.swissinfo.org
|
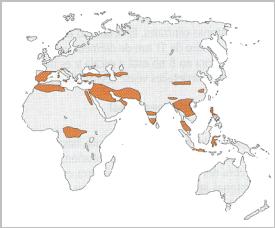
Imatge
15: Localització de
les diferents formes de la malaltia
Genética: Texto y Atlas
|
|
Degut a les migracions continues de
població d’unes àrees a unes altres,
virtualment no existeix cap país al món on
la talassèmia no afecti a un tant per cent dels
seus habitants, és més, està calculat
que un 10% del total dels habitants del món, són
portadors d’un gen talassèmic.
La beta talassèmia es troba en
molts països en especial a la conca del Mediterrani,
la Xina, el SE asiàtic i com hem dit anteriorment
a l’Àfrica.
A Espanya, la beta talassèmia minor és
relativament freqüent, especialment en algunes zones
del litoral mediterrani. Per altra banda, la beta talassèmia
mayor és infreqüent ja que la probabilitat
més gran d’aparició d’aquest
trastorn es produeix quan tots dos membres de la parella
són heterozigòtics.
L’alfa talassèmia és l’alteració genètica
més estesa al món, i en especial a l’Àsia, la conca
Mediterrània i els països africans.
A Espanya, l’alfa talassèmia
es considera, al contrari que la beta talassèmia,
molt escassa,
Símptomes generals de la
malaltia
Cada variant de la malaltia compta amb
els seus propis símptomes; dels més lleus
quan parlem de la talassèmiaminor,
als més greus quan es tracta de la talassèmia
mayor.
La llista següent enumera tots els
símptomes que pateixen els malalts talassèmics
en general, sense diferenciar els diferents graus de la
malaltia:
- Astènia (fatiga)- Dificultat
respiratòria
- Pal·lidesa
- Anèmia
- Insomni
- Inflament de peus i mans
- Danys als òrgans
- Problemes
de creixement
- Risc de taquicàrdies
- Palpitacions
- Bufs
- Cefalées
- Fotòpsies (problema visual)
- Coagulació intravascular
- Deformitats als ossos de la cara
- Icterícia (coloració groguenca
de la pell degut a la bilirrubina)
Característiques microscòpiques
Una de les característiques de
la talassèmia és que els seus glòbuls
vermells pateixen una sèrie de deformacions, ja
que la hemoglobina que contenen és defectuosa. Per
aquesta raó la visió microscòpica
de la sang d’una persona talassèmica és
diferent a la d’una persona sana. Aquests canvis
es fan evidents en l’aspecte dels glòbuls
vermells o eritròcits, ja que són els portadors
de l’hemoglobina, patint una sèrie de deformacions
en comparació als eritròcits d’una
persona sana.
Aquestes cèl·lules són
diferents bàsicament en tres aspectes:
- les seves formes són diferents (poiquilocítics),
- el seu color és més pàl·lid
(hipocròmics),
- i la seva grandària és més reduïda
(microcítics)
Diferents graus de la malaltia...
L’hemoglobina normal en un adult
està formada per quatre grups prostètics,
cada un amb un nucli de ferro, i quatre cadenes proteiques
iguals dos a dos anomenades globina alfa (α) i globina
beta (β).
Un individu normal adult és portador
dels quatre al·lels (dos corresponents a cada pare)
que dicten si aquesta proteïna serà fabricada
correctament o hi haurà algun canvi puntual (segons
les lleis de l’herència).
Segons la globina que es veu afectada,
parlem de diferents tipus de talassèmia:
- α talassèmia: disminució (α+)
o absència (α0) de síntesi de l’alfa
globina.
- β talassèmia: disminució (β+)
o absència (β0) de síntesi de la
beta globina.
- δβ talassèmia:
disminució o absència de les globines
beta i delta.
- Formes rares que produeixen
hemoglobines estructuralment anormals i en petites
quantitats.
L’herència de la beta talassèmia
ens diu que dues persones portadores tenen un 25% de possibilitats
de concebre un fill homozigotic, mentre que en l’alfa
talassèmia és més complicat degut
a que per a sintetitzar aquesta globina alfa, es necessiten
dos gens diferents.
Alfa (α) talassèmia
Els diferents tipus d’alfa talassèmia
depenen del nombre de còpies afectades de les quatre
possibles: |
| GENOTIP |
FENOTIP |
αα
αα |
NORMAL
|
α –
α α |
PORTADOR
SILENT (normal) |
– –
α α |
Tal-1
|
TALASSÈMIA
|
α –
α – |
Tal-2
|
α –
– – |
HbH
|
– –
– – |
HIDROPESÍA
FETAL
|
Taula 1. Taula
amb els diferents genotips de l’α talassèmia
La
taula està ordenada de tal
manera, que els al·lels de cada cromosoma estan
disposats de forma horitzontal.
Per exemple, veiem que
en l’α talassèmia 1, tots dos al·lels
que no duen la informació es troben en el mateix
cromosoma.
- α talassèmia
0 o portador silent:
Es produeix per la delecció d’un
dels quatre al·lels i l’anomenem silent
pel fet que no presenta clars símptomes clínics
ni al laboratori. Calen estudis més precisos
i tècniques
de biologia molecular.
- α talassèmia
1 i 2:
L’α tal-1 es caracteritza
per la desaparició de dues còpies alfa
del mateix cromosoma, mentre que en l’α tal-2
desapareix una còpia de cada cromosoma. Es tracten
de talassèmies
semblants a la beta talassèmia minor, tot i
que són més difícils de diagnosticar
degut a que no hi ha un augment en l’HbA2 ni
en l’HbF.
Per a diagnosticar-la calen mètodes de biologia
molecular.
No sol provocar problemes de salut importants
però els individus afectats poden patir fortes anèmies,
icterícia, fatiga, migranyes, etc... i transmetre
la malaltia als seus descendents.
Per altra banda, on si que es troben
diferències
més notables és en l’aspecte dels eritròcits.
Aquests són més petits i el seu color és
més pàl·lid, i la seva quantitat d’hemoglobina és
més aviat baixa.
Aquests pacients generalment no necessiten
un tractament especial tot i que esporàdicament
poden ser tractats amb àcid fòl·lic
(Acfol) donat que per la seva moderada anèmia
crònica en ocasions pot tenir una despesa extra
d’aquesta vitamina. Per altra banda, aquestes persones
mai han de ser tractades amb ferro, tot i que acostumen
a mostrar dèficit d’aquest element, donat
que el ferro no beneficia al pacient i a la llarga inclòs
pot resultar perillós.
L’α tal-1 es troba en major
mesura en el continent africà, mentre que l’α tal-2
la trobem en major nombre a l’Àsia.
- hemoglobinopatia h(HbH)
En
aquest cas, l’individu hereta
per part d’un pare l’α talassèmia
2 (delecció d’un al·lel) i α talassèmia
1 per part de l’altre pare (delecció de dos
al·lels del mateix cromosoma), per tant no funcionen
3 dels 4 al·lels.
L’excés de cadenes beta forma
l’HbH (β4) es comporta com una hemoglobina inestable.
Els pacients amb afecció d’HbH pateixen fortes
anèmies durant tota la vida amb estigmes i exacerbacions
per infeccions. En general les persones afectades per aquest
tipus de talassèmia arriben fins a l’edat
adulta tot i que acostumen a morir vora els vint anys
degut, generalment, a atacs de cor.
Aquesta malaltia, per primera vegada
va ser descrita en una família d’origen xinès
i en individus d’origen grec. Posteriorment s’ha
demostrat que aquesta afecció és particularment
freqüent a la regió mediterrània (en
especial a les illes), i al sud-est asiàtic. Es
calcula que cada any naixen uns 14.000 nens afectats amb
l’HbH, els quals no tenen una mitjana de vida
superior als 50 anys.
El tractament per a aquesta malaltia
són
les transfusions freqüents de sang per a alleujar
les conseqüències de les anèmies i,
en alguns casos, se’ls pot arribar a extraure la
melsa quan les anèmies són molt severes.
Cal dir que l’alfa talassèmia por agreujar-se
degut a infeccions, alguns fàrmacs com ara les sulfonamides
i sulfones, les fenacetines, la vitamina K, etc., que poden
agreujar l’anèmia i produir icterícia.
A més a més, de forma periòdica
seran tractats amb àcid fòl·lic.
- talassèmia α mayor
(hidropesía fetal)
És el
cas més sever, ja
que l’individu no obté cap dels quatre gens
de part dels seus pares per a la producció de la
globina α. Això és degut a que hereta
de cada pare l’α talassèmia 2.
Està caracteritzada per no sintetitzar
ni l’HbA ni l’HbF, sintetitzant en canvi l’Hb
Barts, amb quatre cadenes gamma (g4) que és moderadament
insoluble i sorprenentment funciona durant l’estat
fetal.
Aquesta situació és incompatible
amb la vida, ja que la mort del fetus es produeix a les
30 o 40 setmanes de gestació o bé, poc tems
després del naixement. Tot i això s’han
descrit casos en els que els fetus han nascut de forma
prematura i han pogut sobreviure un cert temps amb contínues
transfusions de sang.
Els fetus afectats, pateixen anèmia
greu, insuficiència cardíaca i acumulació de
líquids; són petits, edematosos i presenten
distensió abdominal.
L’hidropesía fetal cal intentar
diagnosticar-la durant l’època prenatal amb
la tècnica del PCR o Southern Blot d’ADN procedent
del líquid amniòtic o d’una biòpsia
de les vellositats coriòniques. El tractament consisteix
en transfusions intrauterines i hi ha algun cas excepcional
en què es practica un trasplantament de medul·la òssia
cap als 21 mesos.
Aquest tipus de talassèmia, no
té cap tipus de tractament que pugui assegurar la
longevitat de la persona i s’acostuma a trobar en
el SE asiàtic.
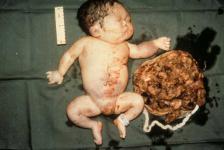
Imatge
17: Nadó que
pateix alfa talassèmia mayor (hidropesía
fetal).
http://www.macmed.ttuhsc.edu
|
Beta (β) talassèmia
Els diferents tipus de beta talassèmia són
els següents:
| GENOTIP |
FENOTIP |
β+ β+ |
NORMAL |
β+ β0 β0
heterozigota |
TALASSÈMIA
MINOR |
Homozigota
(no dependent de transfusions) |
TALASSÈMIA
INTERMITJA |
β0 β0 β0
homozigota |
TALASSÈMIA
MAYOR |
Taula 2. Taula
amb els diferents genotips de la beta talassèmia
Cada
dada que trobem en la columna del genotip (β+
o β0 pertany a cada
un dels al·lels heretats per un individu.
|
-
β-talassèmia minor (tret
talassèmic).
La talassèmia heterozigotica beta
apareix quan solament està afectada una de las dues
còpies del gen que codifica la cadena. Es tracta
de la forma més comú de talassèmia
en què els seus individus sónheterozigotics.
Després de l’α talassèmia 2 ,
es tracta de la forma talassèmica en la que l’expressivitat
clínica és més lleu o menys severa,
fet pel qual també se la coneix com a tret
talassèmic. Es
caracteritza per patir anèmies amb certa periodicitat,
icterícia i astènia o fatiga, amb descens
del VCM i amb nivells de l’Hb A2 gairebé el
doble del normal. Els seus eritròcits, com en altres
tipus de talassèmies, presenten un aspecte microcític
i hipocròmic.
La talassèmia minor està present
des del naixement, roman durant tota la vida i pot transmetre’s
de pares a fills, és a dir, és tracta d’un
trastorn hereditari.
- β talassèmia
intermitja
Es tracta de la forma talassèmica
en la que l’expressivitat clínica és
troba entre la corresponent a les formes de talassèmia
minor i mayor. Són pacients heterozigotics que presenten
una simptomatologia més severa que els que pateixen
beta talassèmia minor. Genèticament, són
pacients, els progenitors dels quals solen ser heterozigòtics.
La possibilitat de que els pares siguin homozigòtics és
molt baixa degut a que són persones amb una esperança
de vida molt baixa i amb una no molt bona qualitat
de vida.
En general, els pacients pateixen anèmia
moderada o intensa, tot i que pràcticament mai requereixen
transfusions. A més, algunes persones, pateixen úlceres
a les extremitats i retràs en el desenvolupament.
Es pot dir, que es tracta d’un síndrome hemolític
crònic, amb pal·lidesa, icterícia
intermitent, esplenomegalia (engrossiment de la melsa),
inflor de peus, astènia i alteracions òssies
moderades.
- talassèmia β mayor
(Anèmia de Cooley)
 |
És el tipus de talassèmia
beta més greu ja que no es sintetitzen
cadenes beta. Els infants semblen sans
al nàixer, però durant
el primer i segon any de vida no tenen
apetit, el seu creixement és lent,
presenten icterícia i es tornen
apàtics i irritables. A partir
dels tres anys comencen a patir una distensió abdominal
força aparent, degut a que el
cor, la melsa i el fetge, es comencen
a inflar degut a l’acumulació de
ferro en els seus teixits.
Els seus ossos es tornen dèbils
i trencadissos. Degut a l’expansió dels
pòmuls i la melaoclusió dental
els pacients amb anèmia de Cooley,
tenen uns trets facials mongòlics.
Les metacarpofalanges i les metarsofalangestenen
una forma rectangular i convexa, les
vèrtebres i els óssos
llargs es fracturen o bé hi
ha una expansió paravertebral
i hi ha risc de compressió.
Altres vegades, existeix la possibilitat
de la fusió prematura de les
epífisis dels ossos llargs que
condicionen l’escurçament
dels braços.
El retràs del desenvolupament
corporal, és atribuït a les
intenses anèmies que pateixen.
A més a més, aquesta falta
de desenvolupament, s’acompanya
del retràs de la producció de
certes hormones que tenen a veure amb
el creixement, i per tant, és
una altra característica, el retràs
en l’arribada de la pubertat. |
| |
 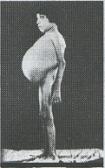  Imatge
18: Infants
que pateixen beta talassèmia
mayor.
Imatge
18: Infants
que pateixen beta talassèmia
mayor.
Genética:
Texto y Atlas |
|
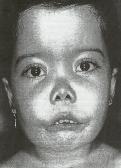 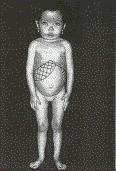
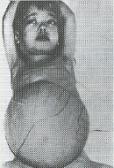
Imatge 19: Nena
amb anèmia de Cooley o beta
talassèmia mayor. Són
característics els trets mongòlics,
la escassa estatura i la presència
d’una intensa esplenomegalia
(distensió abdominal).
Hematología
clínica (→ veure
bibliografia)
|
En relació amb els òrgans
interns, aquests pacients, són més propensos
a patir un gran nombre d’infeccions, entre les
quals, i una de les més importants, es troba
l’hepatitis
B, i és un altre factor, que junt amb les fortes
arítmies, angines de pit i bufs que pateixen
aquests pacients, fan que la seva vida no superi, per
mitja, la vintena d’anys tot i que amb les noves
tècniques
tractament, la seva esperança de vida cada dia
pot ser més alta.
|
|
Moltes de les complicacions de la talassèmica mayor
poden evitar-se sometent als nens a transfusions de sang
freqüents (cada 2 o 3 setmanes) destinades a mantenir
el nivell d’hemoglobina en els seus valors gairebé normals.
Aquest tractament millora el creixement i el benestar general
del nen i, per lo general, prevé la insuficiència
cardíaca i la deformació dels óssos.
Lamentablement, la pràctica repetida de transfusions
de sang deriva en una acumulació de ferro en el
cos que pot afectar el cor, el fetge, el pàncreas
i altres òrgans. Un fàrmac (deferoxamina),
anomenada quelant de ferro, s’uneix a aquest
element i ajuda al cos a desfer-se de l’excés.
Generalment, s’administra durant tota la nit utilitzant
una bomba mecànica que reparteix la droga per sota
de la pell mentre el nen dorm. Una altra tècnica
que remou els dipòsits de ferro i facilita l’excreció és
el consum de vitamina C, que es consumeix
diàriament
en les quantitats que el pacient necessita.
Els individus amb talassèmia beta mayor que reben
transfusions de sang freqüents i quelant de
ferro viuen fins als 30 o 40 anys, i fins i tot més.
Donat que el tractament intensiu amb aquests quelants reben
va començar a practicar-se a la dècada dels
60, i és possible que els nous estudis demostrin
que els individus tractats podran viure encara més.
S’ha aconseguit curar més de 1.000 pacients
amb talassèmia beta a nivell mundial mitjançant
trasplantaments de medul·la òssia, malgrat
això, aquest tipus de tractament tan sols poden
practicar-se en una petita quantitat de pacients amb un
donant de medul·la compatible. Amés a més,
el procediment del trasplantament té un cert
risc i fins i tot pot provocar la mort del pacient.
|
 |
|

Última
actualització:
|
